Theoretical and computational studies of electron conduction across single molecules
Understanding electron transfer (ET) across molecules is very important in two major areas.
In molecular electronics, its impact will be to bring closer the goal of designed functionality in addition to the requirement for robust and reproducible self-assembly. It would also be essential to making the molecular structures integrable with current technology. ET processes also play a fundamental role in driving many important biological functions, e.g., respiration, photosynthesis, biosynthesis. These depend on the ET between the cofactors in proteins and so are dependent on the environment around the proteins.
We are interested in getting a better understanding of the factors that influence electron conduction across single molecules by
- examining the different models that have been and are currently being used;
- direct computation of the electrical conduction using a number of different approaches;
- constructing models of molecular conduction that takes account of molecular vibrations.
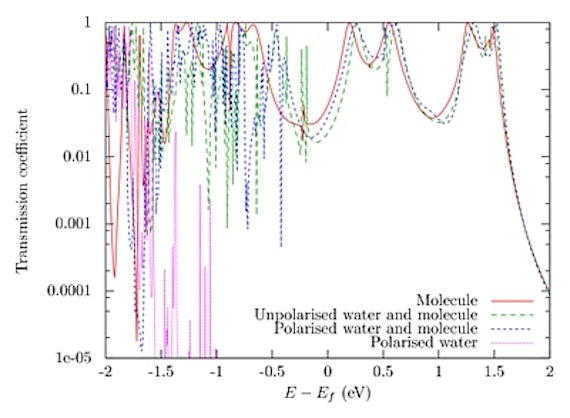
Density functional theory (DFT) approaches have been used in many such studies. However, the size of the molecular systems of interest take them beyond the capability of these methods. We have developed computer codes for carrying out semi-empirical electronic structure and transport calculations on very large structures inaccessible to ab initio techniques.
Activities
Our program of research is continue with our investigations by focussing on small proteins with a view to determining the factors that influence conductivity. Because of the special factors that need to be considered when dealing with proteins, we intend to develop theories that include vibronic effects that have been shown to be important in the study of ET in bio-molecules. This will enable us to study thermal effects on the conduction pathways in these systems.

Publications
- Jones, G. , Elliott, M. and Matthai, C. C. 2012. Electrical conduction along porphyrin wires using the self-consistent extended-Huckel and non-equilibrium Green's function methods. MRS Proceedings 1414 , pp.20-25. (10.1557/opl.2012.138)